


No, AlphaFold has not completely solved protein folding
Biology is hard. Yes, even for AI.
AlphaFold has captured the imagination of people outside biology to an extent not normally seen for a technical tool of computational biology. No tech bro in Silicon Valley has an opinion on HMMER, BLAST, or FoldX, or their potential impact on the future of humanity. But when it comes to AlphaFold everybody knows exactly what it is and does: AlphaFold is an AI tool that has completely solved the protein folding problem. You just give it any arbitrary amino acid sequence and it’ll give you the corresponding protein structure, no questions asked. And, due to its success, all remaining open problems in biology are now similarly within reach of AI-based approaches. We’re maybe a decade away from no longer needing trained biologists or experimental labs because AI knows the answers. The end of disease. No more world hunger. De-extinction of long-lost megafauna.
And yet, working biologists are often more reserved.
To be clear: AlphaFold is an amazing piece of software, and it has definitely moved the needle in computational protein structure prediction. The Nobel Prize was justified. But has AlphaFold completely solved the protein folding problem? Let’s discuss.
We should start by defining what the problem is. In the words of Ken Dill, renowned biophysicist and elected member of the National Academy of Sciences:
The protein folding problem is the question of how a protein’s amino acid sequence dictates its three-dimensional atomic structure.—Dill et al., Annu. Rev. Biophys. 2008.
There’s a lot of complexity hidden under this simple statement, as proteins are dynamic entities and don’t necessarily have a single, well-defined structure. For now though let’s ignore these subtleties and assume we’re interested in finding the ground state, which is the structure the protein spends the majority of its time in.1 Has AlphaFold2 solved this more restricted problem?
What’s the official word?
Google Deep Mind and the European Molecular Biology Laboratory (EMBL) have partnered to provide a public database of 200 million protein structures predicted by AlphaFold. As part of this effort, they have a FAQ page that lists various known limitations of AlphaFold. Let me quote a few statements from this FAQ:
AlphaFold has not been validated for predicting the effect of mutations. In particular, AlphaFold is not expected to produce an unfolded protein structure given a sequence containing a destabilising point mutation.
In other words, AlphaFold cannot reliably tell us whether a protein has a well-defined ground state or not. AlphaFold will typically give us a structure even if the protein does not actually reliably fold at all.
Where a protein is known to have multiple conformations, AlphaFold usually only produces one of them. The output conformation cannot be reliably controlled.
Some proteins spend extended amounts of time in a handful of different conformations. For example, they may switch back and forth between two different states, possibly driven by external inputs such as the presence or absence of binding partners. AlphaFold doesn’t know about this dynamic behavior and will just arbitrarily produce one of the conformations. We have no idea about which one we’ll get and we can’t reliably influence what AlphaFold will do.
For regions that are intrinsically disordered or unstructured in isolation AlphaFold is expected to produce a low-confidence prediction […] AlphaFold may be of use in identifying such regions, but the prediction makes no statement about the relative likelihood of different conformations (it is not a sample from the Boltzmann distribution).
Some proteins have parts that are intrinsically disordered, which means these parts will never fold into a well defined structure. They just flop around. An example could be a protein consisting of two structured domains connected by a flexible linker. AlphaFold can fold the structured domains but doesn’t know what to do with the linker. If you’re lucky, it will assign a low confidence score to that part of the protein. You will get some weird-looking structure that you shouldn’t take too seriously. AlphaFold will definitely not be able to tell you how the linker region behaves dynamically over time.
I could just end this post here. We have our answer. There are several things that AlphaFold can’t do that we would consider to be part of the protein folding problem, even under the more narrow definition where we’re only interested in the protein’s ground state. AlphaFold has not completely solved protein folding.
But, I’m on a roll, so let’s keep going.
Biology is weird
One aspect of biology that is difficult to fathom for anybody who hasn’t devoted decades of their life to studying it is how many weird edge cases exist that any theory or computational method has to be able to handle gracefully. In the context of protein structure prediction, one of my favorite examples is a protein sequence that can stably fold into two different structures, depending on only a single amino acid substitution. In other words, one version of the sequence folds into one structure, and if we make just a single change to the sequence it folds into an entirely different structure. This is quite remarkable, because normally the rule of thumb in protein biochemistry is that two sequences that share as few as 30-50% of their amino acids will generally fold into the same structure. Here, two sequences are 98% similar and yet fold into different structures!
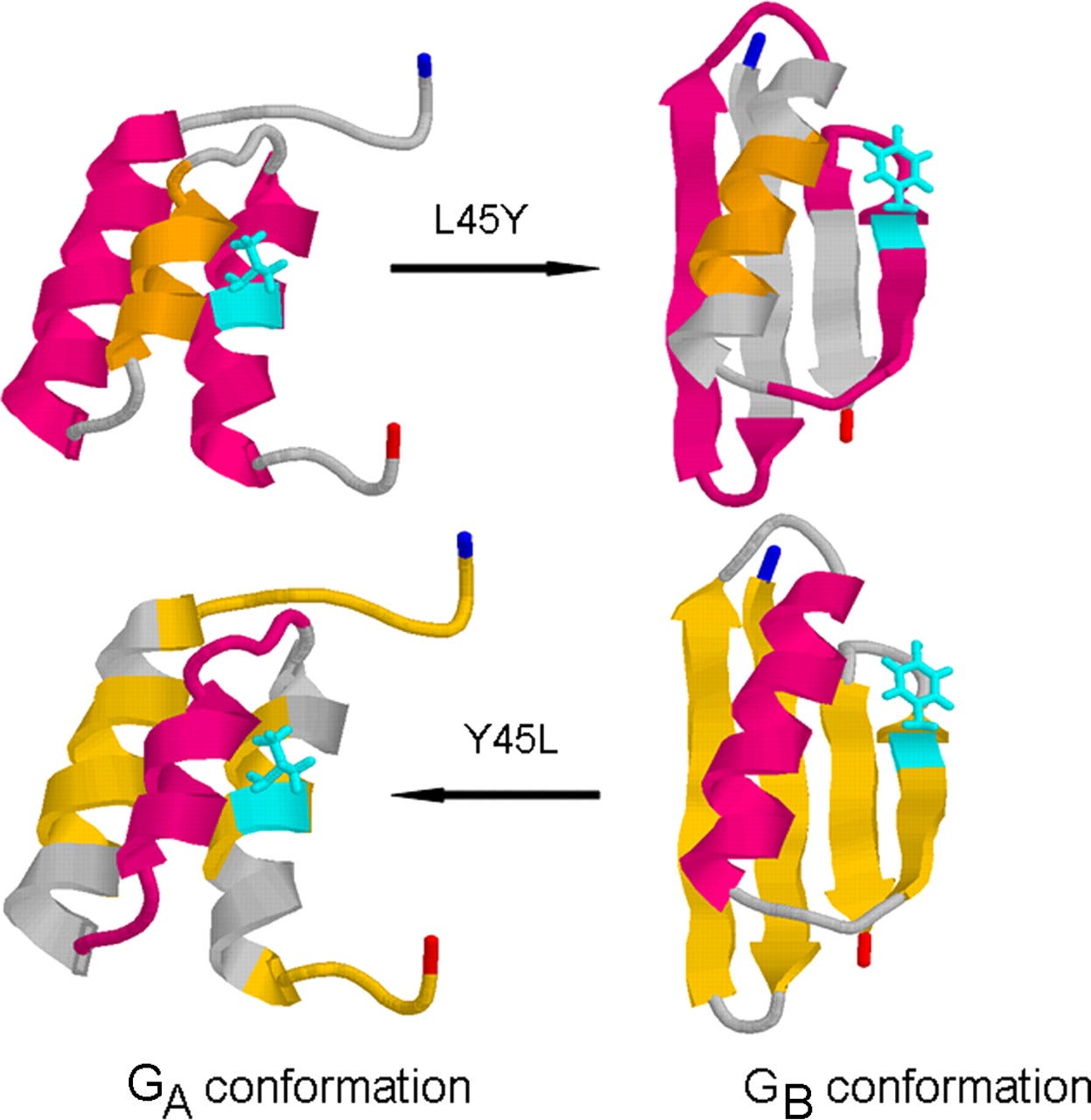
Such proteins are kryptonite for AlphaFold. They undermine all its assumptions. A key component of the AlphaFold algorithm is to build a large alignment of known, closely related protein sequences and then find patterns of covariation in these alignments. (Sites that covary in an alignment are likely in physical contact, and this knowledge of likely physical contacts then constrains and informs the AlphaFold algorithm.) If two closely related sequences fold into entirely different structures then this approach falls apart. The multiple sequence alignment will contain a mix of two different covariation patterns corresponding to two different structures. At best, this will result in AlphaFold being biased towards one of the two structures.3 At worst, this will confuse AlphaFold sufficiently that it gets neither structure right.
Systematic benchmarks uncover AlphaFold’s strengths and weaknesses
There are various systematic assessments of AlphaFold’s performance. I want to focus on a few that highlight important failure modes. For example, one study assessed AlphaFold’s ability to predict the effect of individual point mutations. The above example of fold switching upon mutation notwithstanding, most point mutations do not meaningfully alter a protein’s ground state. However, they do affect the stability of said ground state, up and to the point where the protein no longer folds at all. Thus, when it comes to point mutations, the main question is what effect they have on the protein’s stability. It turns out AlphaFold cannot assess protein stability:
We found a very weak or no correlation between AlphaFold output metrics and change of protein stability or fluorescence. Our results imply that AlphaFold may not be immediately applied to other problems or applications in protein folding.—Pak et al., PLoS One 2023

Another study systematically explored AlphaFold’s ability to predict the structures of fold-switching proteins. These are proteins that are known to exist in at least two different conformations, for example in the presence or absence of a binding partner. AlphaFold performed surprisingly poorly on these proteins:
AF’s inability to accurately predict and score the multiple experimentally determined conformations of fold switchers suggests that the model has more to learn about protein energy landscapes. Complete understanding would enable AF to accurately predict both conformations of fold-switching proteins and their relative frequencies. AF2’s lack of energetic understanding is evidenced by (1) its inability to predict ~65% of fold switchers likely in its training set, (2) its inability to predict >85% of fold switchers outside of its training set, (3) its failure to accurately score models of alternative conformations, and (4) the inability of its structure module to distinguish between low and high energy conformations.—Chakravarty et al., Nat. Commun. 2024
Overall, they found convincing evidence that AlphaFold frequently recalls memorized structures rather than predicting novel structures from scratch:
We found that AF2’s predictive success for some fold-switching proteins results from “memorization” of structures in its training set. This memorization can be so strong that AF2 uses it to inform predictions instead of coevolutionary information detected by its Evoformer.—Chakravarty et al., Nat. Commun. 2024
Memorization works great in the vast majority of cases where evolutionarily related sequences fold into nearly identical structures, but it’ll be confidently wrong in the cases that go against this general pattern.
Finally, I found a review article that covers a lot of the available literature and does a good job describing AlphaFold’s strengths and weaknesses. Let me quote one paragraph that can serve as an excellent overall summary of the topic:
AlphaFold and other machine learning based structure prediction software represents a giant leap forward in our understanding of protein function and structures. However, they are not yet “one-sized fits all” solutions to the protein structure prediction problem. Current implementations of AF2 can provide highly accurate working models for most rigid, well-folded globular proteins, but may have issues predicting other classes of proteins.—Agarwal & McShan, Nat. Chem. Biol. 2024
Going beyond the ground state
Biophysicists have historically focused on predicting the ground-state conformation of proteins because this problem is both sufficiently difficult and sufficiently important that it is a perfectly reasonable starting point. But in the long run, we will have to do better. We will have to be able to predict the entire Boltzmann ensemble of a protein. We will have to be able to predict how a protein dynamically moves through these states. We will have to be able to predict whether a ground state even exists. We will also have to be able to predict how the Boltzmann ensemble changes under different conditions, such as the presence or absence of specific solvents, cofactors, or binding partners.
In time, I am confident we will solve all of these issues. But none of this will be quick or easy. And it’s not immediately clear to me that AI will be the right approach for all remaining problems, though I acknowledge it may be. The ground-state prediction problem was poised for success with AI approaches, in part because the biomedical research community had accumulated databases of hundreds of thousands of experimentally determined ground-state structures and hundreds of millions of protein sequences. In addition, we had known for a long time that modeling new structures based on existing ones (referred to as homology modeling) was a successful strategy. And, predicting structures from sequence covariation was shown to be successful almost a decade before the first release of AlphaFold. What AlphaFold did better than any other prior tool was to combine all the available data and these two distinct methods of structure prediction (homology modeling and covariation) into a single, seamless algorithm. But in a way, this was low-hanging fruit, the culmination of decades of work by an army of dedicated scientists.
It’s not obvious to me that we have similar low-hanging fruit at this time to tackle the next set of problems. For example, we don’t have a massive database of protein dynamics. We can’t even measure protein dynamics all that well.4 We will make progress on all these issues going forward, and our tools for protein structure prediction are only going to get better, but I’m not worried about protein biophysicists and molecular biologists running out of stuff to do in my lifetime.
The ground state is also called the native state or the native conformation or simply the structure or the fold. Yes, that’s a lot of different words that all mean the same thing.
Strictly speaking, there is AlphaFold, AlphaFold 2, and AlphaFold 3. I will not distinguish between these versions here because virtually everything I say applies across the board. The original AlphaFold was quite impressive but still had substantial limitations. AlphaFold 2 was a major improvement over the original AlphaFold. For simple proteins that fold stably into a globular structure AlphaFold 2 will generally give you the right answer. AlphaFold 3 added support for other molecule types beyond proteins, such as DNA, RNA, or small molecules.
If one of the two structures is overrepresented in the sequence alignment, for whatever reason, maybe just due to random chance, then AlphaFold may systematically favor it.
Yes, I know about NMR. It’s a useful tool for exploring protein dynamics. It also has numerous limitations that mean we can’t measure all the data we’d like to have.
Good post. My post was the one that kicked this discussion off, so I'll bring my comments from the thread here too.
On that thread, I made this point to halvorz:
> How much of protein folding that people care about falls into the category that AF is good at? I feel like “naturally evolved, stably folding proteins that exist in a single major conformation” was the majority of what people cared about 10 years ago...In general research will always push forward to the areas where we know the least, so I'm not super surprised that you as a researcher spend most of your time in exactly the areas that existing tools struggle most with.
So here. You made the claim on that thread that, on a scale from "0 to 10, where 0 is nothing and 10 is the entire problem is solved. Before AlphaFold we were at 1. Now we’re at 1.5. Yes, it’s a huge improvement over 1, but there’s still 8.5 points to go."
Obviously this is subjective and there's no hard-and-fast rule about these things, but I think I just disagree with you on this.
AF has provided a database of 200 million proteins, and that too arguably the ones in the categories that we most care about. Yes, it is true that there are edge cases -- I spent enough time in molecular biology to know this -- so perhaps it is wrong to say that AF 'completely solved' protein folding. But also, before AF2, we only had 200k protein structures. So AF improved our protein coverage not by 10x, or 100x, but 1000x.
I think you are right that AF does not give us explainability, but that to me is shifting the goal posts. In 2015, it was assumed that being able to predict protein structure from sequence necessarily meant you had a good understanding of the dynamics. In 2025, we realize that you can figure out the former without the latter. It turns out for most *applications* we dont really care about the latter.
And of course, I think very few people now believe that the path to 'solving' the 'rest' of protein folding is going to come from techniques like xray crystallography or whatever. Even though you hint otherwise, imo the protein configurations that we currently *can't* solve -- the limitations of AF -- are more likely to be solved by other AI tools or applications of AI tools, than they are to be solved by a deep rules-based theoretical understanding of electron charges and physics, at least in the short term. So, fine, AF may not have 'completely solved' protein folding semantically. But I think what most people are grasping at when they say things like "AF has completely solved protein folding" is that AI has revolutionized this field that was previously seen as totally intractable, as it has done in many other fields in similar settings.
Have you heard of Labbot? I’ve been really impressed by it. It tackles many of the challenges mentioned here, not by predicting structure, but by experimentally tracking how proteins behave in real conditions. Things like conformational changes, aggregation, binding events... even under temperature shifts or pH changes. It’s a great complement to AlphaFold when you need to understand what proteins actually do, not just how they might look.