


Limitations of protein language models applied to viral data
Biology is hard. Yes, even for AI.
Current biological AI models don’t seem to work well for data from viral proteins. Specifically, I’m referring to protein language models applied to the problem of predicting effects of mutations. Protein language models are transformer-based AI models similar to ChatGPT but trained entirely on protein sequences. The most popular such models are the various ESM models developed by the company EvolutionaryScale.
Protein language models have been tremendously successful in many applications. In particular, they excel at transfer-learning applications, where we use the embeddings from these models to predict variant phenotypes. For example, we can make thousands of mutants of a fluorescent protein and use a protein language model to predict which mutations lead to high fluorescence and which do not. Except, when we do this for viral proteins, it doesn’t really work.
My lab has been studying protein language models for a while, and we recently published a systematic benchmark of various models applied to a wide range of different datasets. While the primary purpose of this study was to assess how model performance relates to model size, we made an unexpected discovery along the way: Protein language models just don’t work very well for datasets of viral proteins. I have expanded on this thought some more in a recent PNAS commentary, and I’m reproducing here the figure I prepared for that commentary. (The plotted data is entirely from the benchmarking study though.) I think the difference between non-viral and viral proteins is striking.
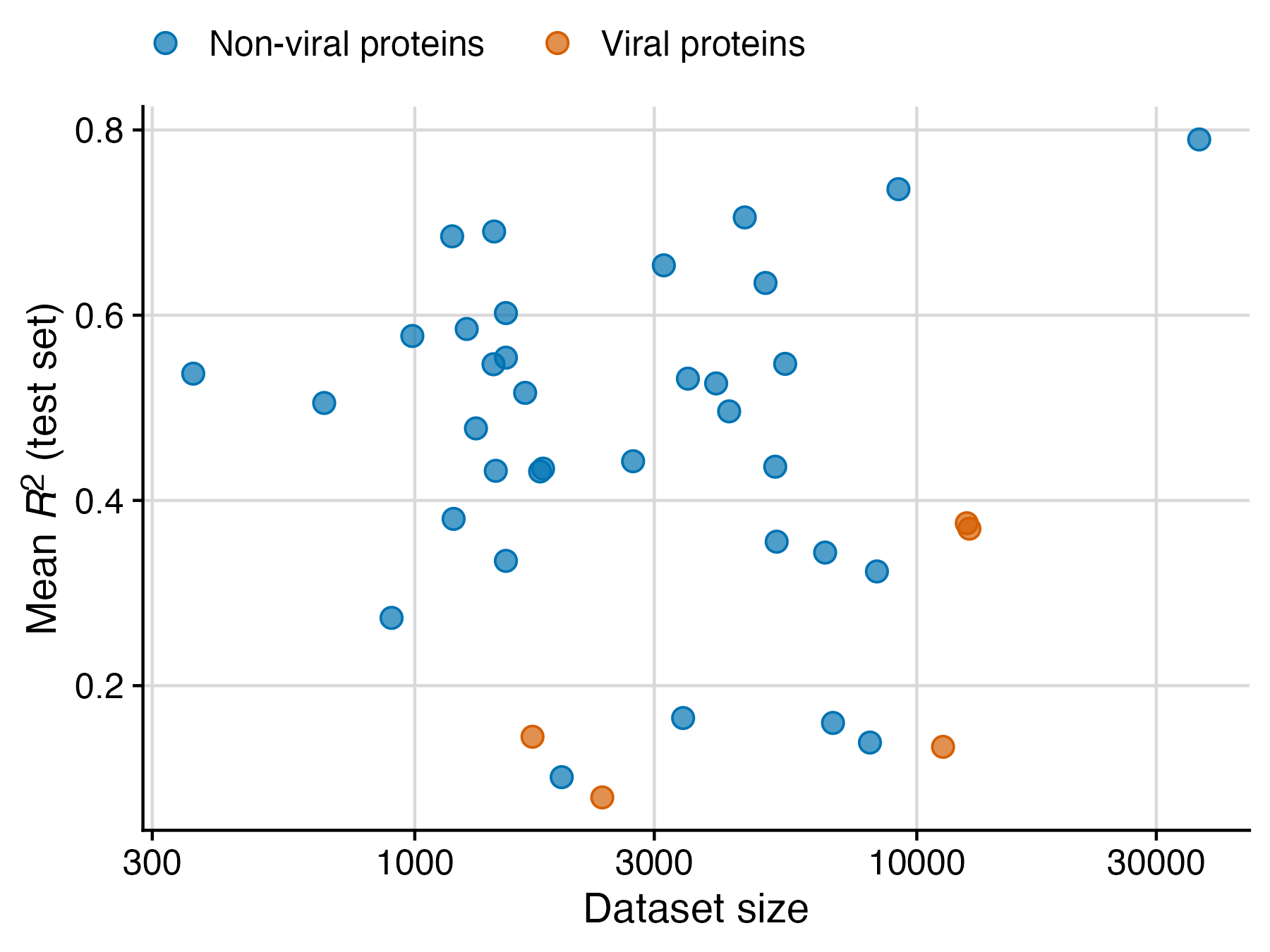
I realize this figure has only five viral datasets. So it’s totally fair if you’re now wondering whether this is just an unfortunate selection of datasets. The answer is no, but I’m not yet in the position to show you. In recent weeks, we have curated many more viral datasets, and the pattern generally remains. To date we have found very few viral datasets that yield an R2 greater than about 0.4, while many non-viral datasets far exceed this value, as you can see from the figure. We’ll publish this data in due course.
What is causing this pattern? In brief, we don’t know. There are two possible causes, not mutually exclusive. First, the models aren’t any good for viral proteins. Second, viral proteins have some property that makes variant effect prediction particularly difficult, even for good models. I suspect both of these causes are to some extent at play. This is a topic of ongoing and future study.
We’re not the only ones who have noticed that protein language models don’t work well for viral data. See for example this paper by Debora Marks’ group, and this paper that fine-tuned a protein language model specifically for viral proteins. But I don’t think this fact is widely acknowledged yet. In fact, others are warning of impending doom, going as far as envisioning a future where AI models will rapidly design dangerous viruses. I understand this is a fast-moving field, and maybe next week somebody will release a language model that works spectacularly well for viral proteins. But as of today, I’m much more concerned about our apparent inability to do good virology research with existing models. Current models simply aren’t performing well enough to give us consistently good results.
Hey Claus, I stumbled on this blog by chance and it turns out that I'm trying out these models for predicting fitness of single point mutation variants of HIV-1. How do you think viral epistatic interactions may be modelled? Because it seems more like a physics problem for me at this point, maybe we need to integrate AI and physics principles at some level?
Do you suspect something inherent in virus-specific evolution pressures? Wondering if a higher viral mutation rate leads to greater genetic drift and more chance of mechanistic diversity, more difficult to make predictions about with less data. If you subdivide by say, RNA viruses, do you see a greater split? Easy way to test this idea would be to compare success with which the protein language models predict, say, the results of IgG somatic hypermutation. Really interesting finding, thanks for the post!